
Publication
- Article : 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002
2017
Ion Transport across Biological Membranes by Carborane-Capped Gold Nanoparticles
Marcin P. Grzelczak*, Stephen P. Danks, Robert C. Klip, Domagoj Belic, Adnana Zaulet, Casper Kunstmann-Olsen, Dan F Bradley, Tatsuya Tsukuda, Clara Viñas, Francesc Teixidor, Jonathan J. Abramson, and Mathias Brust
ACS Nano 11, 12492-12499 (2017) ![]()
Structure Determination of a Water-Soluble 144-Gold Atom Particle at Atomic Resolution by Aberration-Corrected Electron Microscopy
Maia Azubel*, Ai Leen Koh, Kiichirou Koyasu, Tatsuya Tsukuda, and Roger David Kornberg
ACS Nano 11, 11866-11871 (2017)![]()
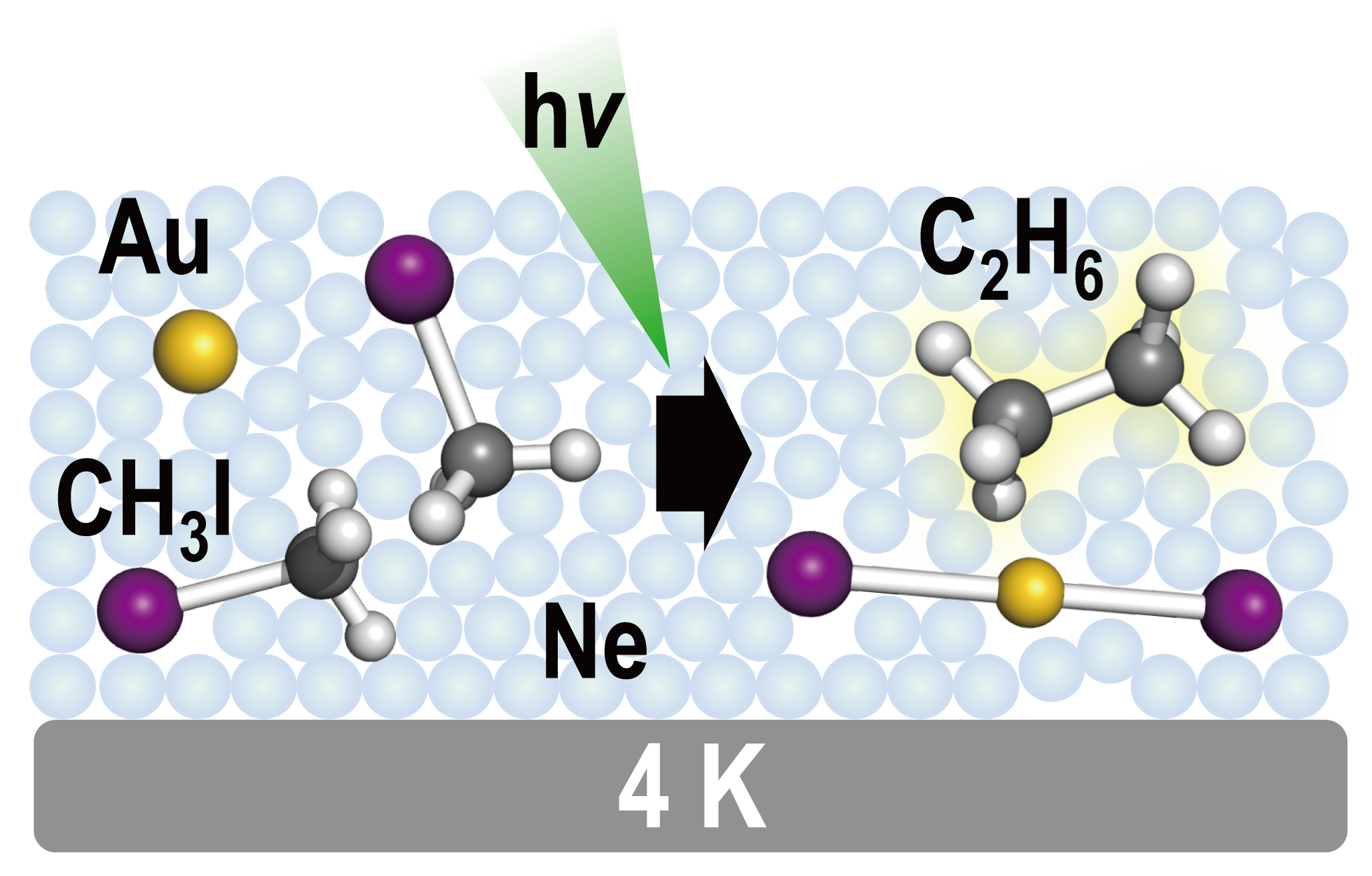
Photoassisted Homocoupling of Methyl Iodide Mediated by Atomic Gold in Low-Temperature Neon Matrix
Satoru Muramatsu, Xuan Wu, Mohua Chen, Mingfei Zhou*, and Tatsuya Tsukuda*
J. Phys. Chem. A, 121, 8408-8413 (2017).![]()
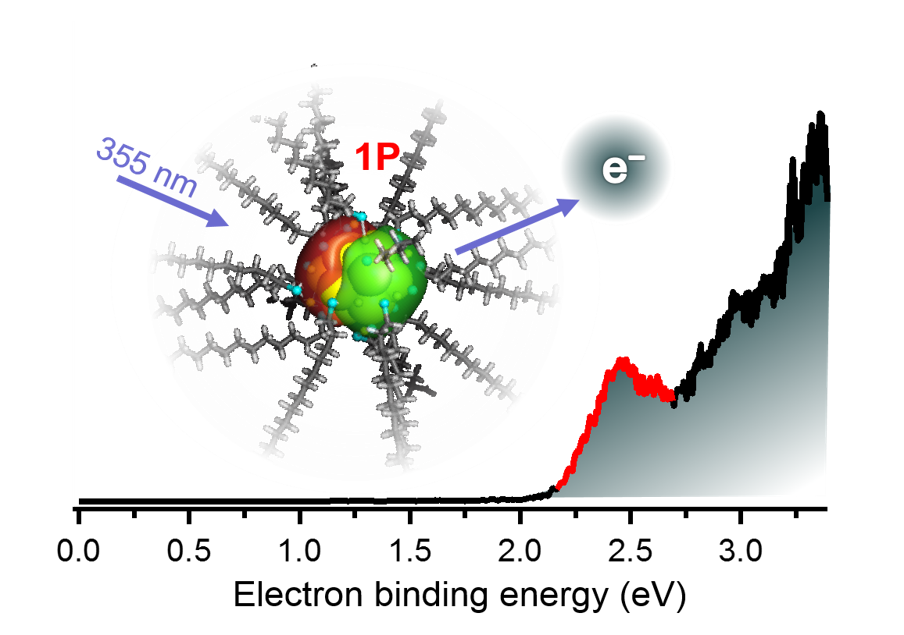
Anion Photoelectron Spectroscopy of Free [Au25(SC12H25)18]−
Keisuke Hirata, Keishiro Yamashita, Satoru Muramatsu, Shinjiro Takano, Keijiro Ohshimo, Toshiyuki Azuma, Ryuzo Nakanishi, Takashi Nagata, Seiji Yamazoe, Kiichirou Koyasu and Tatsuya Tsukuda*
Nanoscale, 9, 13409-13412 (2017).![]()
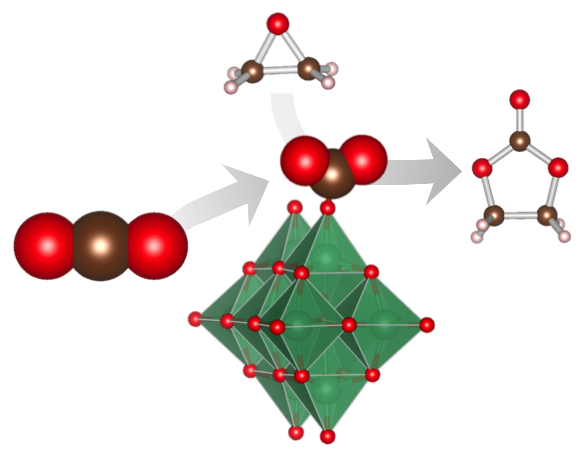
Lewis Base Catalytic Properties of [Nb10O28]6− for CO2 Fixation to Epoxide: Kinetic and Theoretical Studies
Shun Hayashi, Seiji Yamazoe*, Kiichirou Koyasu, and Tatsuya Tsukuda*
Chem. Asian J. 12, 1635-1640 (2017).![]()
Selected in the Spotligihts on our sister journals of Angewandte Chimie International Edition
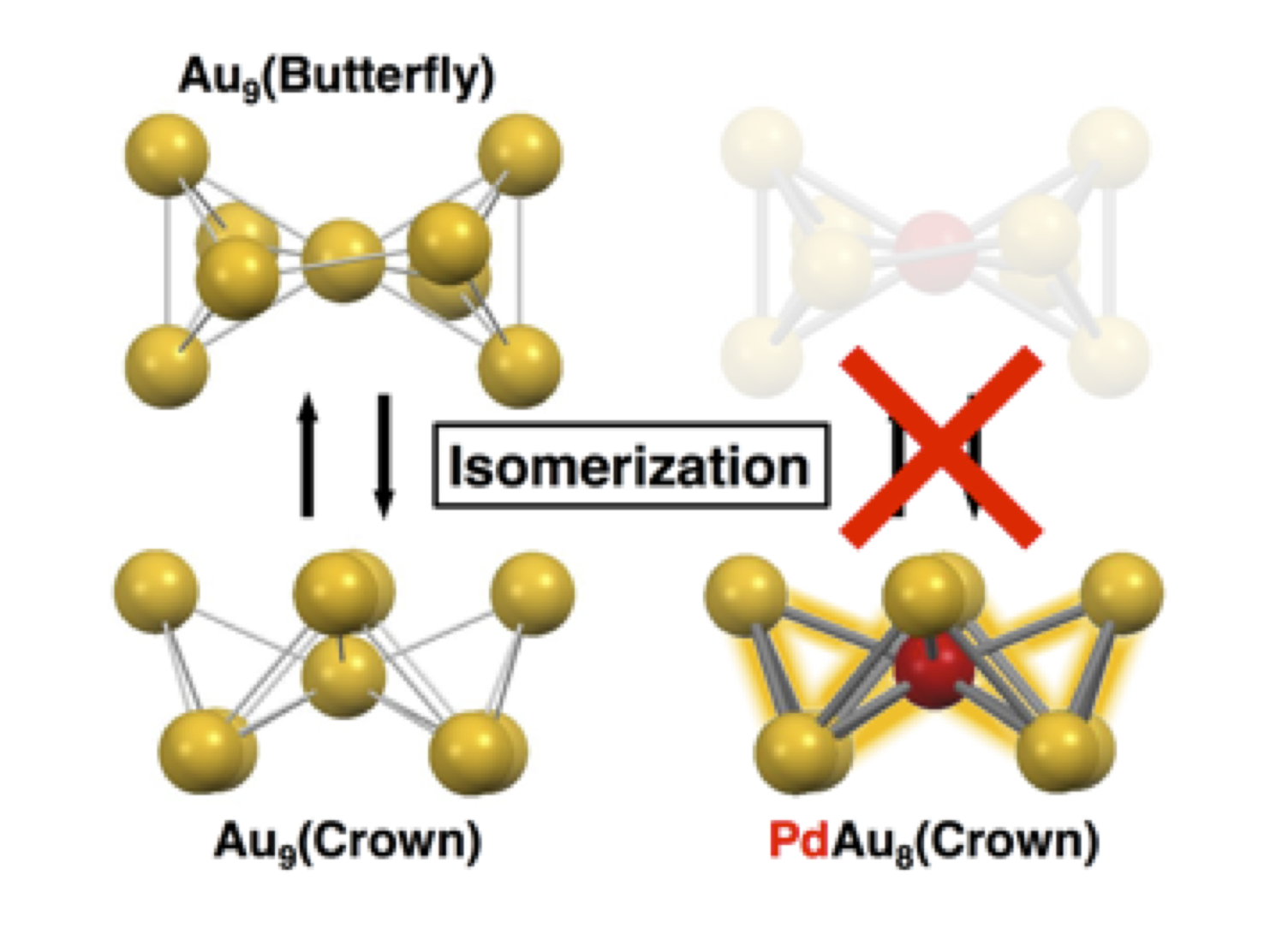
Suppressing Isomerization of Phosphine–Protected Au9 Cluster by Bond Stiffening Induced by Single Pd Atom Substitution
Seiji Yamazoe, Shota Matsuo, Satoru Muramatsu, Shinjiro Takano, Kiyofumi Nitta, and Tatsuya Tsukuda*
Inorg. Chem. 56, 8319-8325 (2017).![]()
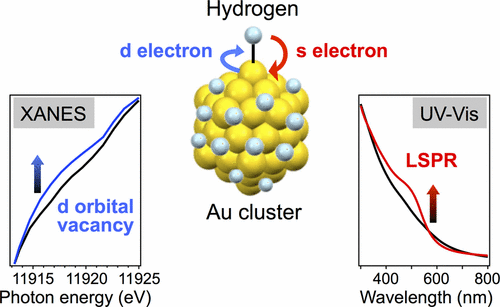
Hydrogen-Mediated Electron Doping of Gold Clusters as Revealed by In Situ X-ray and UV-Vis Absorption Spectroscopy
Ryo Ishida, Shun hayashi, Seiji Yamazoe, Kazuo Kato, and Tatsuya Tsukuda*
J. Phys. Chem. Lett, 8, 2368-2372 (2017).![]()
Selected as ACS Editors' Choice
Selected in Spotlights of JPCL.
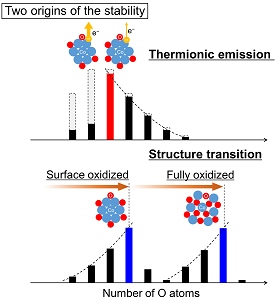
Observation and the Origin of Magic Compositions of ConOm− Formed in Oxidation of Cobalt Cluster Anions
Ryohei Tomihara, Kiichirou Koyasu, and Tatsuya Tsukuda*
J. Phys. Chem. C, 121 (20), 10957-10963 (2017)![]()
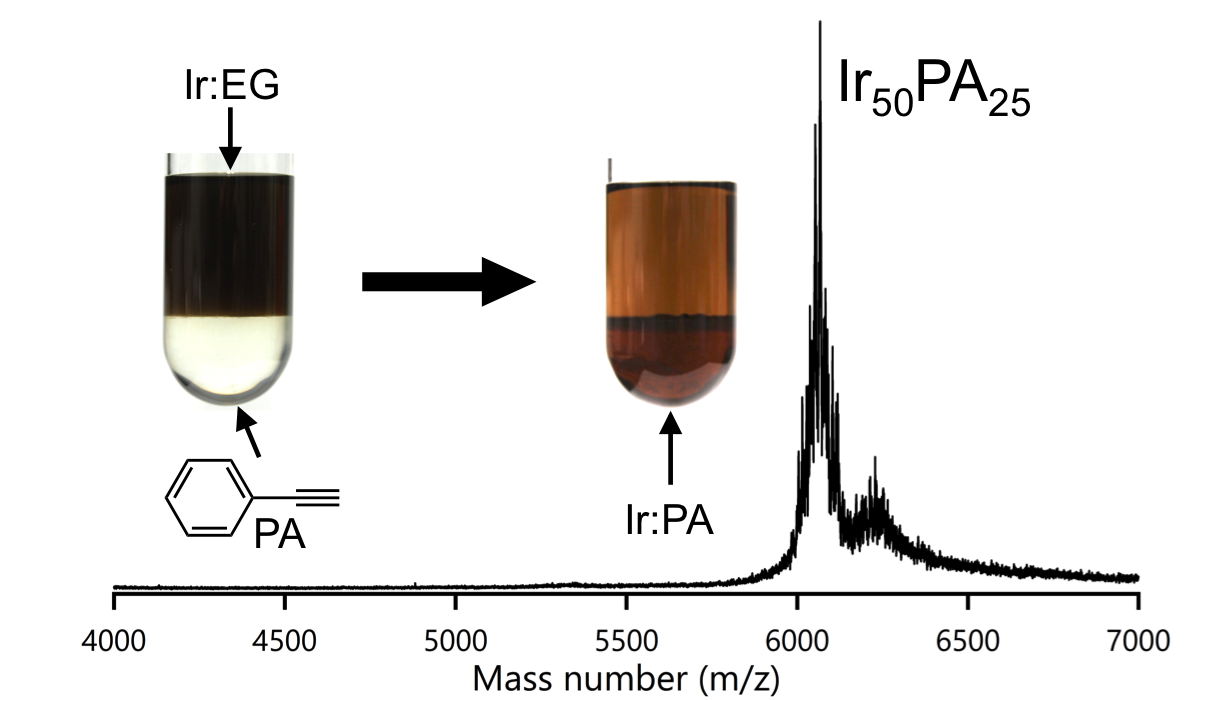
Monodisperse Iridium Clusters Protected by Phenylacetylene: Implication for Size-Dependent Evolution of Binding Sites
Hiroki Yamamoto, Prasenjit Maity, Ryo Takahata, Seiji Yamazoe,
Kiichirou Koyasu, Wataru Kurashige, Yuichi Negishi, and Tatsuya Tsukuda*
J. Phys. Chem. C, 121 (20), 10936-10941 (2017)![]()
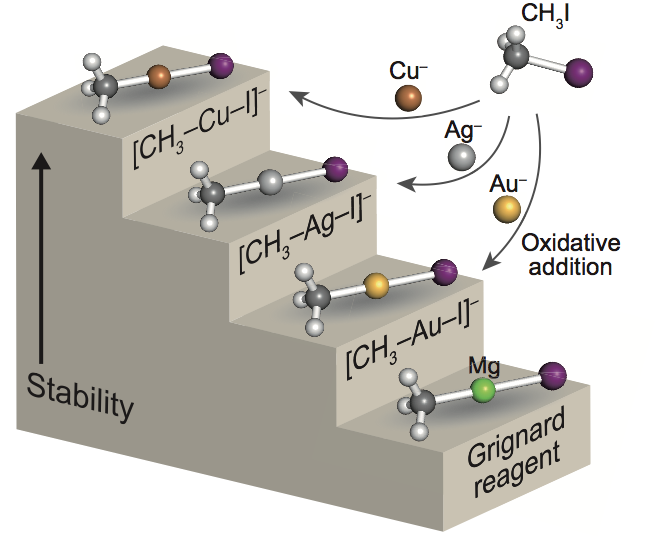
Formation of Grignard Reagent-like Complex [CH3−M−I]− via Oxidative Addition of CH3I on Coinage Metal Anions M− (M = Cu, Ag, Au) in the Gas Phase
Satoru Muramatsu, Kiichirou Koyasu, and Tatsuya Tsukuda*
Chem. Lett. 46, 676-679 (2017).![]()
Structural Model of Ultrathin Gold Nanorods Based on High-Resolution Transmission Electron Microscopy: Twinned 1D Oligomers of Cuboctahedrons
Ryo Takahata, Seiji Yamazoe, Kiichirou Koyasu, and Tatsuya Tsukuda*
J. Phys. Chem. C, 121 (20), 10942-10947 (2017)![]()

A Gold Superatom with 10 Electrons in Au13(PPh3)8(p-SC6H4CO2H)3
Shinjiro Takano, Seiji Yamazoe, and Tatsuya Tsukuda*
APL. Mater. 5, 053402 (2017).![]()